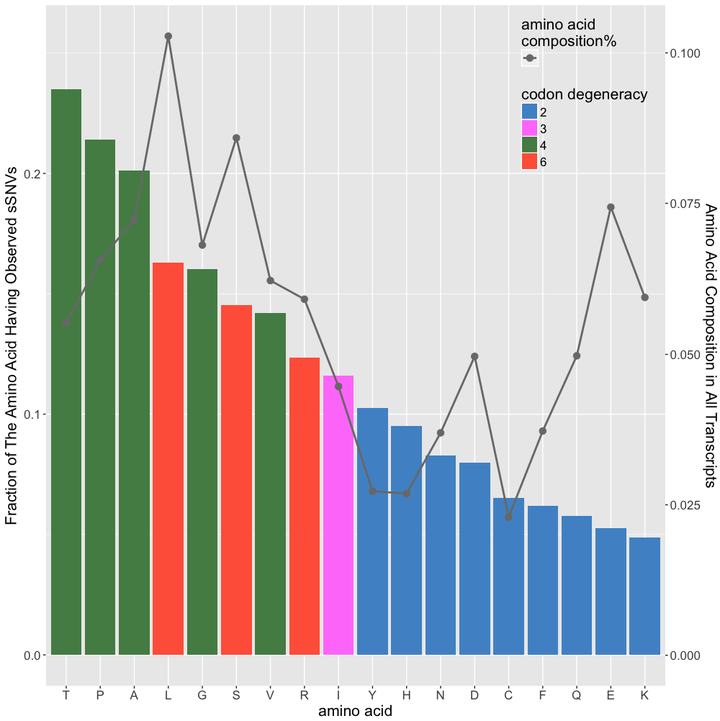
 distribution of amino acid among all observed sSNVs
distribution of amino acid among all observed sSNVs
Decoding the effects of synonymous variants
Synonymous variants are the genetic variants within coding regions that do not alter the amino acid in the protein sequence, e.g., CCC->CCT does not change the amino acid proline. Compared to non-synonymous variants (i.e., variants changing the amino acid), the effects of synonymous variants are often overlooked. However, accumulating evidences have shown that synonymous variants can have large biological effects and even be causative to many diseases. That is why some people referred the effects of synonymous variants as the “the sound of silence”.
Our goal is to develop a prediction tool based on machine learning to evaluate the effects of synonymous variants. Instead of retrieving data from online databases (which, in our opionion, have fundamental limitations), we collected/generated our data based on the assumption that evolution results already harbor the information we need to differentiate deleterious variants from neutral variants. Further, we collected/calculated a number of features for the variants in various aspects, including codon bias, bicodon pattern, codon autocorrelation, mRNA stability, gene expression level, tRNA supply-demand estimate, regulatory factors, splice sites, etc.
We are performing extensive feature selection, not just for the model building, but also to explore the biological importances of the features. In the meantime, after trying a few machine learning algorithms, we are training a deep neural network to see if we can surpass the previous performances. The ultimate goal of this project is to integrate this prediction tool for synonymous variants into our lab’s larger pipeline to predict the predisposition of certain diseases from genomic sequencing data.
Zishuo Zeng
PhD Student
Synonymous variants have the right to remain silent, but they choose not to.