 Improving variant effect prediction with position types
Improving variant effect prediction with position types
Creating functional maps of protein sequences
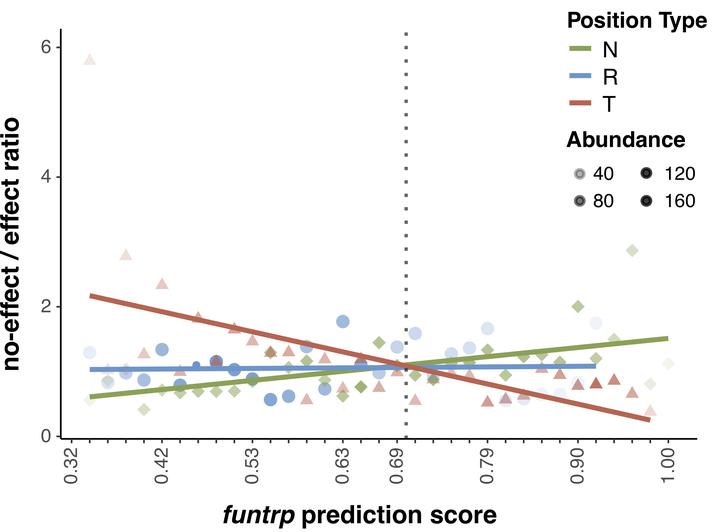
Evaluating the impact of non-synonymous genetic variants is essential for uncovering disease associations. Understanding the corresponding changes in sequence can further facilitate synthetic protein design and stability assessments. Despite continuous efforts in the field, little improvement in performance has been observed in recent years. One reasons for this might be that most approaches exploit similar sets of gene/protein features for model development, e.g. sequence conservation. While high levels of conservation clearly highlight residues essential for protein activity, much of the in vivo observable variation is arguably weaker in its impact and, thus, requires evaluation of a higher level of resolution. We developed function Neutral/Toggle/Rheostat predictor (funtrp) to classify protein sequence positions based on the expected range of mutational impacts: Neutral (mostly no/weak effects), Rheostat (range of effects; i.e. functional tuning), or Toggle (mostly strong effects). Three conclusions of our work are most salient: (i) position types do not correlate strongly with familiar protein features such as conservation or protein disorder; (ii) position type distribution varies across different enzyme classes; (iii) position types reflect experimentally derived functional effects, improving performance of existing variant effect predictors. This suggests that future predictors would greatly benefit from incorporating funtrp functional maps as additional feature.
Maximilian Miller
PostDoctoral Associate
improving variant effect predictions, deciphering metagenomes and creating an efficient service infrastructure