 AVA,DX perfomance compared
AVA,DX perfomance compared
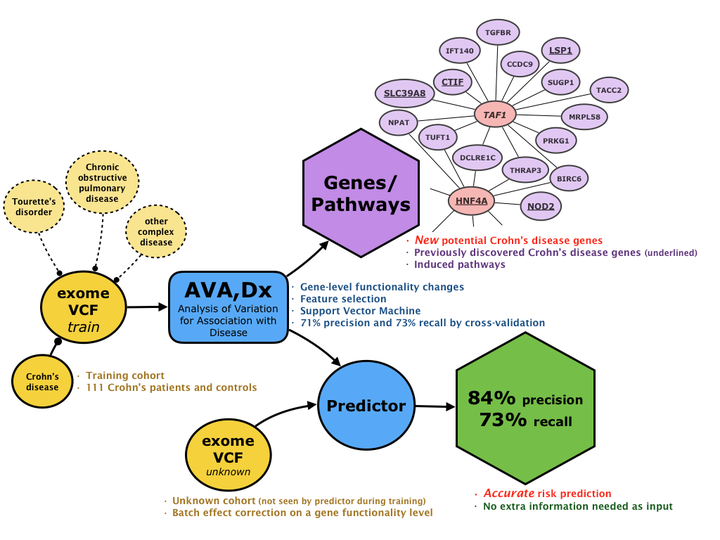
After years of concentrated research efforts, the exact cause of Crohn’s disease (CD) remains unknown. Its accurate diagnosis, however, helps in management and preventing the onset of disease. Genome-wide association studies have identified 241 CD loci, but these carry small log odds ratios and are thus diagnostically uninformative. Here we describe a machine learning method – AVA,Dx (Analysis of Variation for Association with Disease) - that uses exonic variants from whole exome or genome sequencing data to extract CD signal and predict CD status. Using the person-specific coding variation in genes from a panel of only 111 individuals, we built disease-prediction models informative of previously undiscovered disease genes. By additionally accounting for batch effects, we were able to accurately predict CD status for thousands of previously unseen individuals from other panels. AVA,Dx highlighted known CD genes including NOD2 and new potential CD genes. AVA,Dx identified 16% (at strict cutoff) of CD patients at 99% precision and 58% of the patients (at default cutoff) with 82% precision in over 3000 individuals from separately sequenced panels. Larger training panels and additional features, including other types of genetic variants and environmental factors, e.g. human-associated microbiota, may improve model performance. However, the results presented here already position AVA,Dx as both an effective method for revealing pathogenesis pathways and as a CD risk analysis tool, which can improve clinical diagnostic time and accuracy.