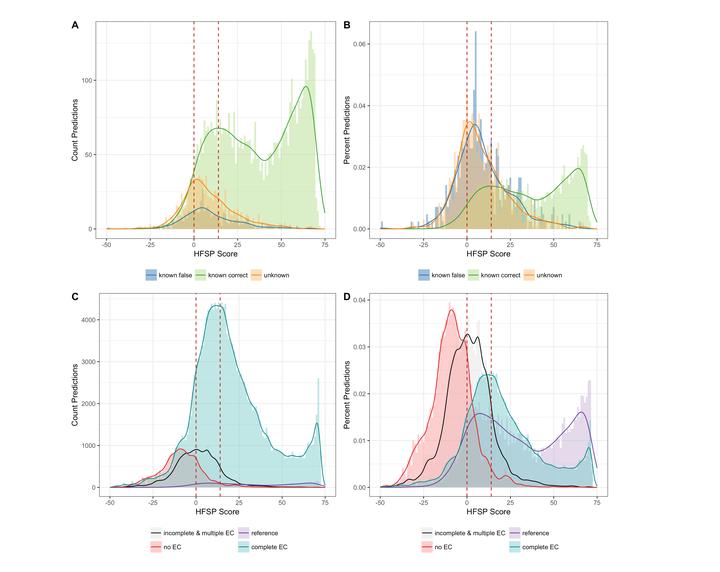
 Newly emerging enzyme functionality difficult to differentiate from incorrect function predictions
Newly emerging enzyme functionality difficult to differentiate from incorrect function predictions
The rapid drop in sequencing costs has produced many more (predicted) protein sequences than can feasibly be functionally annotated with wet-lab experiments. Thus, many computational methods have been developed for this purpose. Most of these methods employ homology-based inference, approximated via sequence alignments, to transfer functional annotations between proteins. The increase in the number of available sequences, however, has drastically increased the search space, thus significantly slowing down alignment methods.
Homology-derived functional similarity of proteins (HFSP) is a novel computational method that uses results of a high-speed alignment algorithm, MMseqs2, to infer functional similarity of proteins on the basis of their alignment length and sequence identity. The method is accurate (85% precision) and fast (more than 40-fold speed increase over state-of-the-art). HFSP can help correct at least a 16% error in legacy curations, even for a resource of as high quality as Swiss-Prot. We suggest HFSP as an ideal resource for large-scale functional annotation efforts.